
TEpredict: predicting T-cell epitopes
This version of TEpredict uses quantitative matrices (QM) based approach. To illustrate the principles underlying MHC-binding prediction we will discuss it on the «RLRPGGKKK» peptide. At first, this peptide is encrypted with sparse encoding: it is represented as an array with shape i*j (20*9) where each nonzero element at position (i,j) codes for aminoacid i at position j:
# A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y
{{0,0,0,0,0,0,0,0,0,0,0,0,0,0,1,0,0,0,0,0},
{0,0,0,0,0,0,0,0,0,1,0,0,0,0,0,0,0,0,0,0},
...
{0,0,0,0,0,0,0,0,1,0,0,0,0,0,0,0,0,0,0,0}}
If aminoacid properties are used to parametrize peptide, selected scale "P" of properties is multiplied with obtained sparse matrix elementwise to produce an array {{P1},{P2},...{P9}} (all zero elements are removed). This array is multiplied with predictive model matrice elementwise. According to the type of selected model, either multiplicative (the majority of models developed by Dr. Raghavas' group) or additive (our models), elements of resulted matrix are either multiplicated or summed up. The resulting score after correction (either multiplication with corrective coefficient or summation with corrective constant according to the type of used model) is compared to the selected threshold; if score is greater than threshold, peptide is considered to be a binder.
Prediction of proteasomal/immunoproteasomal processing works as a filter: peptides, predicted to have no proteasomal cleavage site at their C-terminus are excluded from futher analysis. Default threshold level for these filters equals to 5%, as suggested by authors (Singh and Raghava, 2003). But I recommend you to use 10% threshold.
Models for predicting peptide-TAP binding were taken from the literature sources (Peters et al., 2003; Doytchinova et al., 2004). This option also works as a filter in the current version of TEpredict: peptides predicted to be inefficient TAP-binders are excluded from the further analysis. You can vary stringency of the filter by choosing appropriate threshold values of peptide-TAP binding affinity. For more detailed instructions how to use this function I advise you to read articles, specified above.
 TEpredict GUI.
Here you can select parameters for TEpredict. GUI works only as a
collector of parameters, that should be passed to the working module
through the command line. Programm contains reduced help, but if you
didn't get useful information here, you shouldn't look there: the text
is the same. But I think that TEpredict is so simple that you won't
need tutorials or something like that. If I'm wrong let me know. TEpredict GUI.
Here you can select parameters for TEpredict. GUI works only as a
collector of parameters, that should be passed to the working module
through the command line. Programm contains reduced help, but if you
didn't get useful information here, you shouldn't look there: the text
is the same. But I think that TEpredict is so simple that you won't
need tutorials or something like that. If I'm wrong let me know. TEpredict is able to perform BLAST search for the local similarity of antigens with proteins of interest (e.g. human proteins). All peptides, containing local similarity with human proteins would be excluded from further analysis. For BLASTing TEpredict use local version of BLAST installed on your machine, and your database of proteins, preformatted for BLAST (if you need help for this, mail me or consult your local guru). All parameters for BLAST search should be written in the file, named "BLAST.conf", located in the TEpredicts' directory. Matrix of antigenic similarity of aminoacids is included in TEpredict distributive. If you selected to produce epitope summary, you could use Epitope Selector widget. It helps you to assess expected populational coverage by the selected set of peptides. It even could suggest you a minimal set of peptides covering all MHC alleles used for prediction. An example of results obtained with this option enabled you can see below. |
You could also perform analysis of nonameric peptides, but if you do so, proteasomal/immunoproteasomal filter should be disabled. I recommend you to use only nonumbiguous representation of aminoacids. If TEpredict finds inappropriate letters, results of the predictions made for that peptide could be inadequate. TEpredict is able to analyse of a batch of protein antigens at once.
User should choose threshold level for prediction. Lowering the threshold you are decreasing specificity of prediction and increasing its sensitivity. Default threshold value is optimal for predicting promiscuous MHC-binders. To choose more than one MHC allele for prediction you should use either <Ctrl> or <Shift> keys. You could also choose the number of best scored peptides to be displayed for each chosen MHC allele. By default you'll get all peptides predicted to be binders at the chosen threshold.
You can choose output format to represent prediction results in a way than suits better to your needs:
 1. Epitope map.
Prediction results are outputed as in ProPred1 epitope map.
Capital
letters depict the starting aminoacid of the epitope. Results
of
prediction are saved in HTML format. When cursor is placed on the
starting aminoacid of the epitope the text with information
about
allele and predicted MHC-peptide binding score pop-ups. At the bottom
of the page parameters used for predictions are outlined. 1. Epitope map.
Prediction results are outputed as in ProPred1 epitope map.
Capital
letters depict the starting aminoacid of the epitope. Results
of
prediction are saved in HTML format. When cursor is placed on the
starting aminoacid of the epitope the text with information
about
allele and predicted MHC-peptide binding score pop-ups. At the bottom
of the page parameters used for predictions are outlined. |
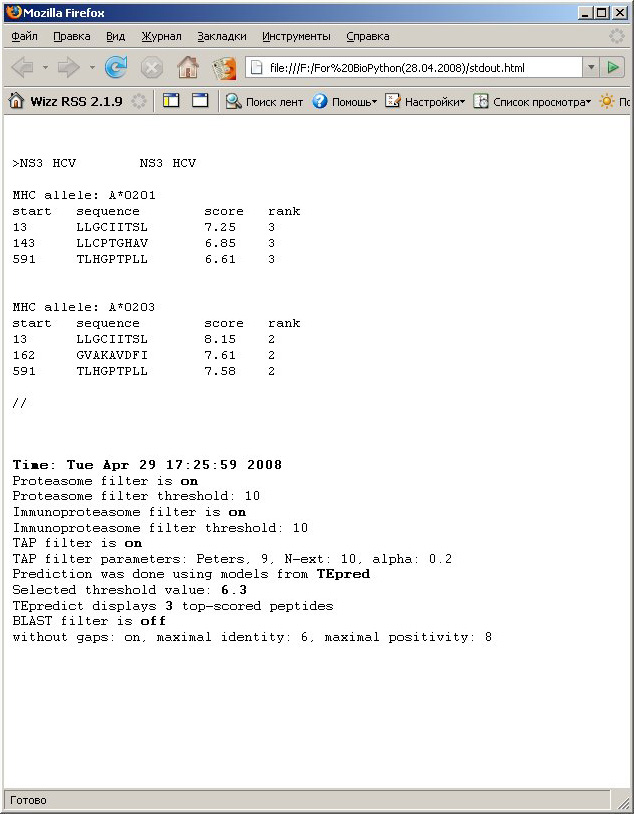 2. Tabulated text
format. Prediction results are displayed as text table with
peptides
sorted in descending order according to predicted MHC-peptide
interaction score. Results of prediction are saved as HTML file with
preformatted text with tab-separated columns. If you save it
as a text file, it's easy to parse and it should be easily processed
with your scripts. 2. Tabulated text
format. Prediction results are displayed as text table with
peptides
sorted in descending order according to predicted MHC-peptide
interaction score. Results of prediction are saved as HTML file with
preformatted text with tab-separated columns. If you save it
as a text file, it's easy to parse and it should be easily processed
with your scripts. |
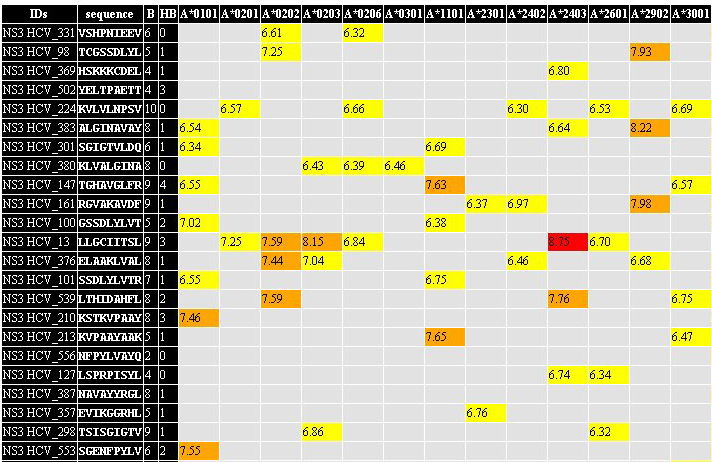 3. HTML matrix
with the summary of the
prediction session.
All predicted epitopes are collected in one summury table. If certain
epitope was found in more then one antigen, all antigens containing
this peptide are outlined in the certain field of the summary table. In
rows, corresponding to the predicted epitopes, the binding
score
(predicted by TEpredict) is outlined
for every MHC allele used for prediction. Color marks of the cells with
prediction results corresponds to peptides' rank. You can
open results
of prediction saved in this format in Excel (or in OpenOffice Calc) and
analyze them as you wish. 3. HTML matrix
with the summary of the
prediction session.
All predicted epitopes are collected in one summury table. If certain
epitope was found in more then one antigen, all antigens containing
this peptide are outlined in the certain field of the summary table. In
rows, corresponding to the predicted epitopes, the binding
score
(predicted by TEpredict) is outlined
for every MHC allele used for prediction. Color marks of the cells with
prediction results corresponds to peptides' rank. You can
open results
of prediction saved in this format in Excel (or in OpenOffice Calc) and
analyze them as you wish. |
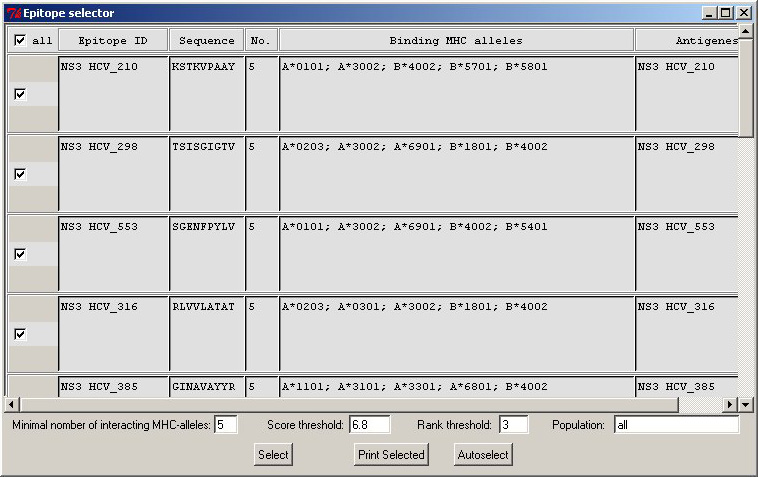 Epitope
Selector widget. It helps you to
assess expected populational coverage by the selected set of peptides.
It even could suggest you a minimal set of peptides covering all MHC
alleles used for prediction. Epitope
Selector widget. It helps you to
assess expected populational coverage by the selected set of peptides.
It even could suggest you a minimal set of peptides covering all MHC
alleles used for prediction. |
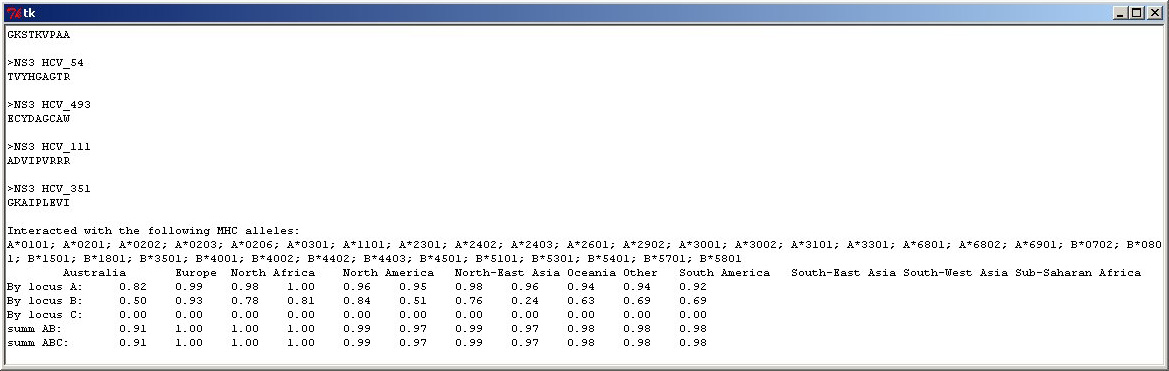 Expected population coverage, predicted with Epitope Selector. |
Enjoy ;)
If you have questions or suggestions (or any other feedback), please mail me.
If you think my code is ugly and you could do much better, you are wellcome! I'm not a programmer, I'm a biologist ;)
References and acknowledgements :)
Abdi, H. (2003) Partial Least Squares (PLS) Regression.
In Lewis-Beck M., Bryman, A., Futing T. (Eds.) Encyclopedia of
Social Sciences Research Methods. Thousand Oaks (CA): Sage,
1528 p. (Thanks him for great introduction in PLS and useful
references)
Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z.,
Miller, W., Lipman, D.J. (1997) Gapped BLAST and PSI-BLAST: a
new generation of protein database search programs. Nucleic
Acids Res., 25, 3389—3402. (Thanks them for the greatest program
for searching local similarity in different proteins)
Bhasin, M., Raghava, G.P. (2004) Prediction of CTL epitopes using
QM, SVM and ANN techniques. Vaccine, 22, 3195—3204. (Thanks to Dr.
Raghavas' group for their articles and great web-servers that
inspaired me to write TEpredict. I'd like to thank them for been so
kind to make their predictive models available through the WWW)
Doytchinova, I., Hemsley, S., Flower, D.R. (2004) Transporter asso-
ciated with antigen processing preselection of peptides binding
to the MHC: a bioinformatic evaluation. J. Immunol., 173, 6813—
6819. (Thanks them for the model for predicting peptide-TAP binding
and for idea to use PLS analysis to construct predictive models)
Kidera, A., Konishi, Y., Oka, M., Ooi, T., Scheraga, H.A. (1985)
Statistical analysis of the physical properties of the 20 natu-
rally occuring amino acids. J. Prot. Chem., 4, 23—55.
(Thanks them for one of the scales, that TEpredict uses to parametrize
peptides)
Liu, W., Meng, X., Xu, Q., Flower, D.R., Li, T. (2006) Quantitative
prediction of mouse class I MHC peptide binding affinity using
support vector machine regression (SVR) models.
BMC Bioinformatics, 31, 7:182. (Thanks them for one of the scales,
that TEpredict uses to parametrize peptides and for great web-server
aimed to T-cell epitope prediction)
Maksyutov, A.Z., Bachinskii, A.G., Bazhan, S.I., Ryzhikov, E.A.,
Maksyutov, Z.A. (2004) Exclusion of HIV epitopes shared with hu-
man proteins is prerequisite for designing safer AIDS vaccines.
J. Clin. Virol., 31, 26—38. (Thanks them for the matrix of aminoacid
antigenic similarity that TEpredict uses to exclude peptides,
shareing local similarity with human proteins)
Mevik, B.-H., Wehrens, R. (2007) The pls Package: Principal Component
and Partial Least Squares Regression in R. Journal of Statistical
Software, 18, 1—24. (Thanks them for the PLS package for R and great
introduction in PLS method)
Peters, B., Bulik, S., Tampe, R., Van Endert, P.M., Holzhutter, H.G.
(2003) Identifying MHC class I epitopes by predicting the TAP
transport efficiency of epitope precursors. J. Immunol., 171,
1741—1749. (Thanks them for the model for predicting peptide-TAP binding)
Peters, B., Sidney, J., Bourne, P., Bui, H.H., Buus, S., Doh, G.,
Fleri, W., Kronenberg, M., Kubo, R., Lund, O., Nemazee, D.,
Ponomarenko, J.V., Sathiamurthy, M., Schoenberger, S., Stewart, S.,
Surko, P., Way, S., Wilson, S., Sette, A. (2005) The immune epitope
database and analysis resource: from vision to blueprint.
PLoS Biol., 3, e91. (Thanks them for the greatest and the most complete
epitope resource created to date)
Singh, H., Raghava, G.P. (2003) ProPred1: prediction of promiscuous MHC
Class-I binding sites. Bioinformatics, 19, 1009—1014. (Thanks to Dr.
Raghavas' group for their articles and great web-servers that
inspaired me to write TEpredict. I also thank them to be so
kind to make their predictive models available through the WWW)
Singh, H., Raghava, G.P.S. (2001) ProPred: prediction of HLA-DR binding
sites. Bioinformatics, 17, 1236—1237. (Thanks to Dr. Raghavas' group
for their articles and great web-servers that inspaired me to write
TEpredict. I also thank them to be so kind to make their predictive
models available through the WWW)
Sing, T., Sander, O., Beerenwinkel, N., Lengauer, T. (2005) ROCR: visu-
alizing classifier performance in R. Bioinformatics, 21, 3940—3941.
(Thanks them for great R package for assessing performance of predictive
models)
And some useful links:
http://rpy.sourceforge.net (RPy makes possible to work with R directly from Python)
http://www.biopython.org (I thank developes for tons of useful code :)
http://www.immuneepitope.org (The greatest epitope resource made ever)
http://www.ncbi.nlm.nih.gov/gv/mhc/ (HLA allele frequences data)
http://www.r-project.org (The powerful statistical environment)
http://www.python.org (The greatest and the most friendly programming language)